将深度学习与分子学习结合:从拓扑、几何和文本角度进行解析
- Chapter 1 简介
- Chapter 2 single-modal and multi-modal pretraining on geometry
- Chapter 3 textual description modality enables more comprehensive tasks
- Chapter 4 总结
- 致谢
Chapter 1 简介
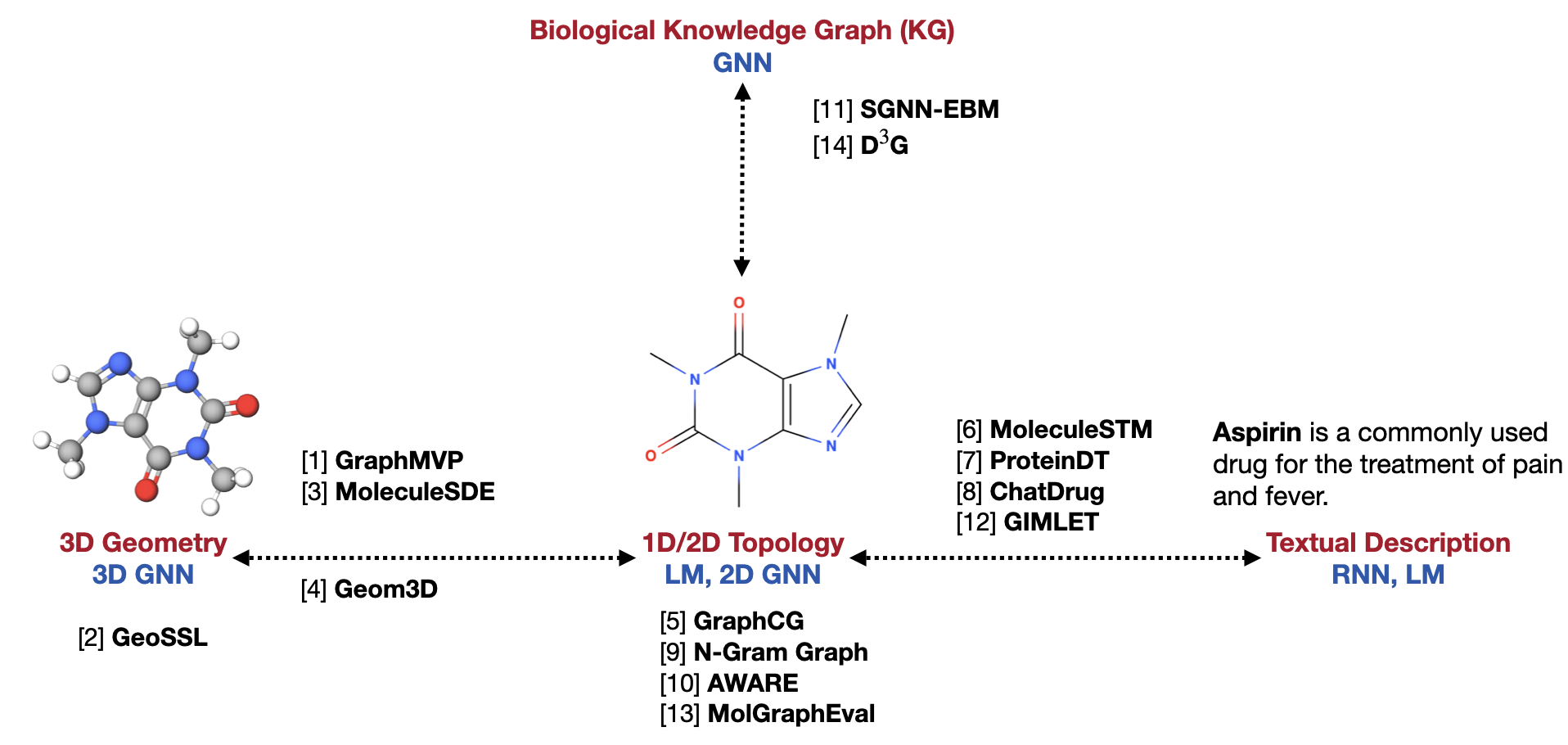
在计算生物、化学、材料领域,伴随着深度学习(DL)的广泛使用,分子的表征(representation)已经成为了最基础的研究问题。一个鲁棒的分子表征,能够支撑丰富的任务。我们团队从研究小分子表征开始研究,是因为它包含了非常丰富的多模态信息,如下图展示的六个模态。
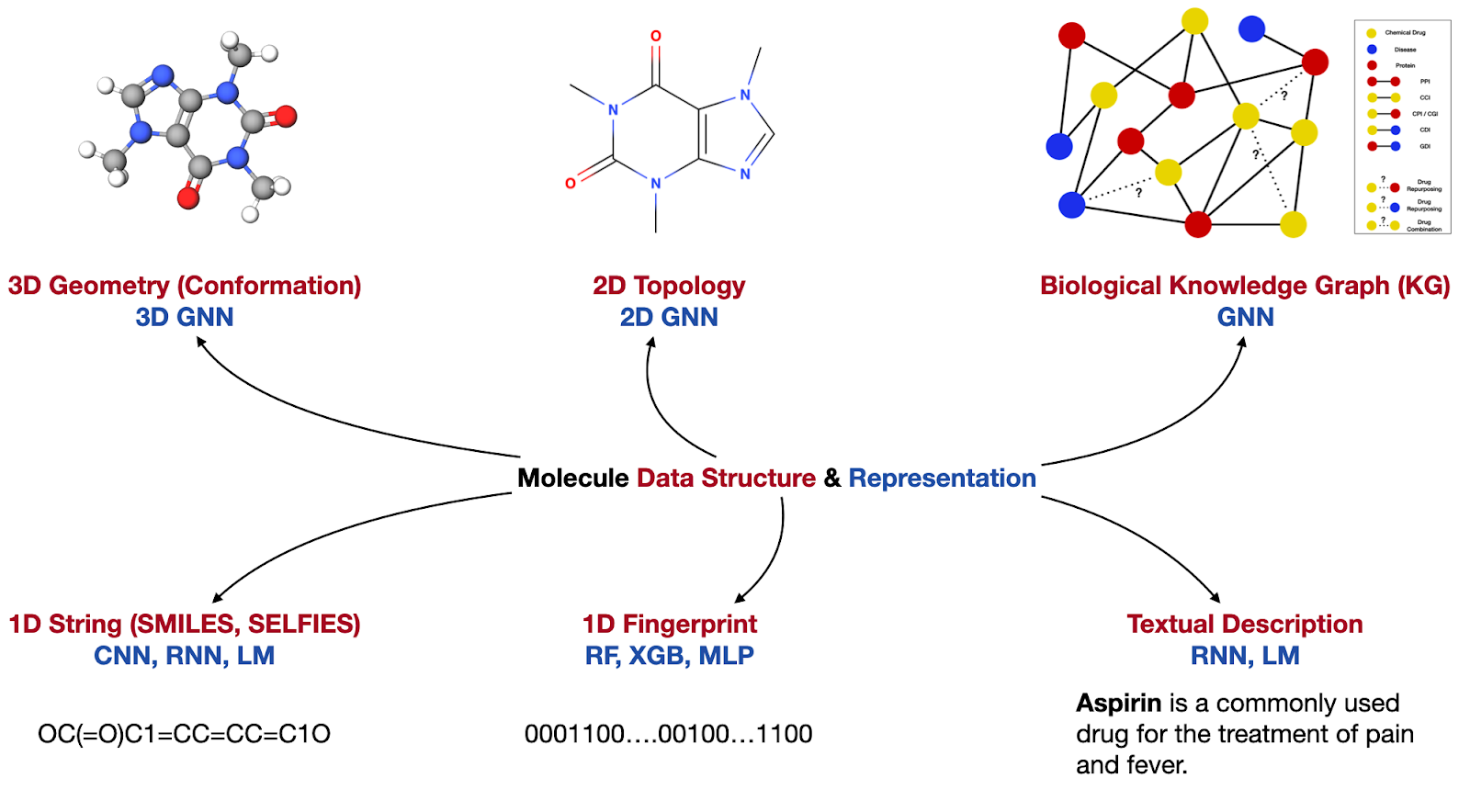
这六种模态又可以分为两大类:3D geometry、2D topology、1D String和1D Fingerprint都是关于小分子的“内部”化学结构表示;而bio KG和textual description则更多的是关于分子的“外部”功能描述。这两大类的模态(即内部和外部模态)是可以相互补充信息。
此外还需要强调的是,对于小分子的内部模态,2D topology和1D String、1D Fingerprint在信息层面等价,主要区别是关于分子图的不同数据结构以及对应的DL表征。但是3D geometry和2D topology在信息层面是很大的区别,并且它们的信息能够互相补充。
围绕以上两点,我们将简单介绍围绕这两种信息互享,介绍两个系列工作:
- geometry相关的多模态任务
- GraphMVP: Pre-training Molecular Graph Representation with 3D Geometry, ICLR 2022
- GeoSSL: Molecular Geometry Pretraining with SE(3)-Invariant Denoising Distance Matching, ICLR 2023
- MoleculeSDE: A Group Symmetric Stochastic Differential Equation Model for Molecule Multi-modal Pretraining, ICML 2023
- Geom3D: Symmetry-Informed Geometric Representation for Molecules, Proteins, and Crystalline Materials, ArXiv 2023
- textual description相关的多模态任务
- GraphCG: Unsupervised Discovery of Steerable Factors in Graphs, NeurIPS Workshop 2022
- MoleculeSTM: Multi-modal Molecule Structure-text Model for Text-based Editing and Retrieval, ArXiv 2022
- ProteinDT: A Text-guided Protein Design Framework, ArXiv 2023
- ChatDrug: ChatGPT-powered Conversational Drug Editing Using Retrieval and Domain Feedback, ArXiv 2023
Chapter 2 single-modal and multi-modal pretraining on geometry
2.1 Geom3D 分子的几何表征benchmark
从数据结构上,分子的本质(稳定态)是一个结构稳定的3D点云。他的表征主要挑战就是如何保证对于旋转、平移等变。现有的工作都是利用了群表示论来进行描述,而我们在Geom3D这个平台中对于现有的工作进行了进一步的整理和总结。现有的geometric modeling可以大致分为如下三类:
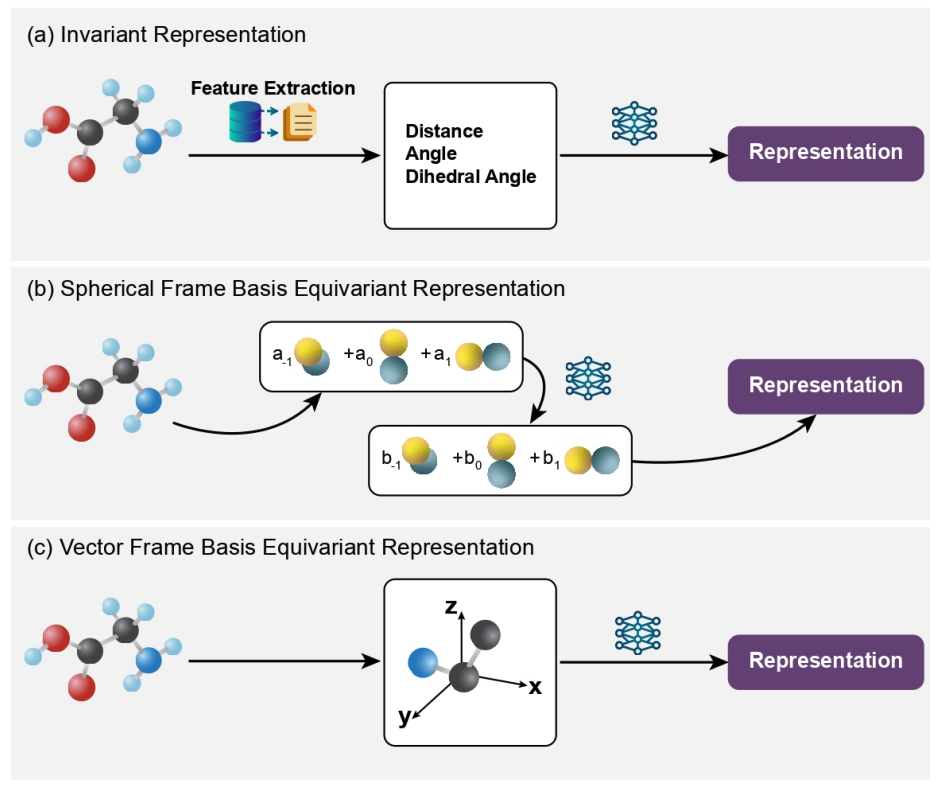
- Invariant modeling,是仅仅只考虑了type-0 feature (比如distance、angle)。
- Equivariant modeling,是除了type-0 feature,还会考虑type-1 feature,这在我们的问题中就是3D coordiante坐标。而为了保证modeling对于SE(3)的等变性,有两种主要的解决思路:
- Equivariant modeling with spherical frame basis 是把相对位置的vector投影到spherical harmonics frame上进行的modeling。它的优势是一种非常泛化的框架,比如可以model更高阶的particle feature (物理问题中使用更多);而缺点则是需要进行tensor product,这个计算复杂度非常高。
- Equivariant modeling with vector frame basis 则更加针对3D coordinates:它是把coordinates投影到了一个vector frame上,然后再对其进行modeling。它的优点是效率比较高,而缺点就是无法对高阶的粒子进行建模。
- 此外我们还想提一下其他的建模方式,比如利用电子轨道特征的OrbNet或者利用李群建模的LieTransformer。
基于这样的设计思路,Geom3D benchmark了如下模型:
- 16个geometric modeling模型
- 14个geometric pretraining模型
- 46个不同geometric tasks,包含了小分子、蛋白质、和材料
- 此外Geom3D还包含了7个1D model和11个2D GNN model。关于2D topology pretraining,我们团队也有一个前序工作,MolGraphEval。感兴趣的朋友欢迎查阅。
下面我们就重点介绍在单一模态和多模态情况下,如何进行geometric pretraining。
2.2 GraphMVP & MoleculeSDE: 2D-3D Pretraining
2.2.1 GraphMVP的结构化数据预训练框架:从最大化互信息到条件概率求和
对于小分子的多模态,我们最先考虑到的就是2D topology和3D geometry。同时对于2D和3D进行预训练,而预训练的思路非常简单,就是最大化2D topology和3D geometry之间的互信息(MI)。但这里有一个难点就是如何对于结构化数据进行MI计算,而GraphMVP最大的贡献是提出来一个关于最大化MI的一个等价形式:
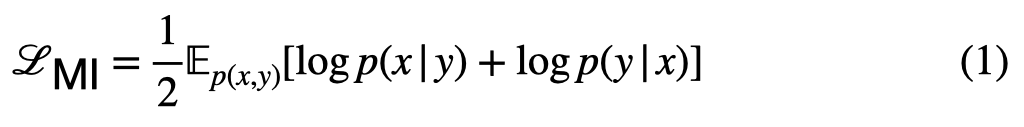
这就把MI maximization问题变成了summation of two conditional log likelihoods (公式1);而且他在这里还有更具体的意义:2D生成3D的条件概率 + 3D生成2D的条件概率。这个形式对于结构化的数据非常友好,因为可以引入EBM这种兼具泛化性、灵活性和强表达能力的概率模型进行估计。
这里我们将结合下面几个工作进行一些延展补充。注:GraphMVP的推导是基于离散情况,GeoSSL还提供了连续版本的推导,且二者最终优化目标一样。感兴趣的朋友可以参考比对两篇文章的附录。
- 首先对于公式(1),我们可以把条件概率用energy-based model (EBM)进行估计,而EBM本身就有非常多的家族方法来求解,比如noise contrastive estimation (NCE)、score matching (SM)、contrastive divergence。
- GraphMVP利用NCE求解,我们叫做EBM-NCE。我们发现EBM-NCE和Jensen-Shannon divergence联系密切。二者的目标函数一样,只是求解的过程和思路不同。而EBM-NCE和其他contrastive self-supervised learning的思路本质都一样:通过构造positive和negative pairs,然后增大positive pair的similarity,并且见效negative pair的similarity。
- 此外EBM还有其他的求解思路,比如score matching (SM),我们当时在做GraphMVP的时候已经意识到了它也是do-able的路,但是第一篇工作来不及详细展开。这个也是指导我们后面做GeoSSL和MoleculeSDE的方法论。
- 其次对于公式(1),我们还可以利用变分方法来估计两个条件概率的evidence lower bound (ELBO)。这个类似VAE和DDPM。
- GraphMVP就首先采用了VAE的形式,提出了variation representation reconstruction (VRR)。VRR是在representation space进行reconstruction (而不是data space),从而有了对ELBO的估计。并且我们发现,non-contrastive self-supervised learning (比如BYOL、SimSiam) 就是VRR的一种特殊情况。
- DDPM也是在优化ELBO,并且它和denoising score matching (DSM) 是非常类似的。它们的区别可以通过Stochastic Differential Equation (SDE)一个统一框架下的两种变形体现。这一点我们在MoleculeSDE中进行了更加详细的解释。
- 此外,对于这几种思路,我们还可以从另一个角度将这些方法进行分类。
- 第一类是NCE (包含了EBM-NCE、InfoNCE、GAN),因为它们本质思路就是把概率估计问题转换为了分类问题,也就是contrastive learning。它是基于data pair来进行distribution estimation。
- 第二类是类似DSM、VRR,目标任务是为了重构某一个data或者data的representation。本质是把概率估计问题转换为了重构问题,也就是generative learning或者reconstruction learning。它是直接基于每一个单独的data point来进行distribution estimation。
2.2.2 基于latent space的GraphMVP
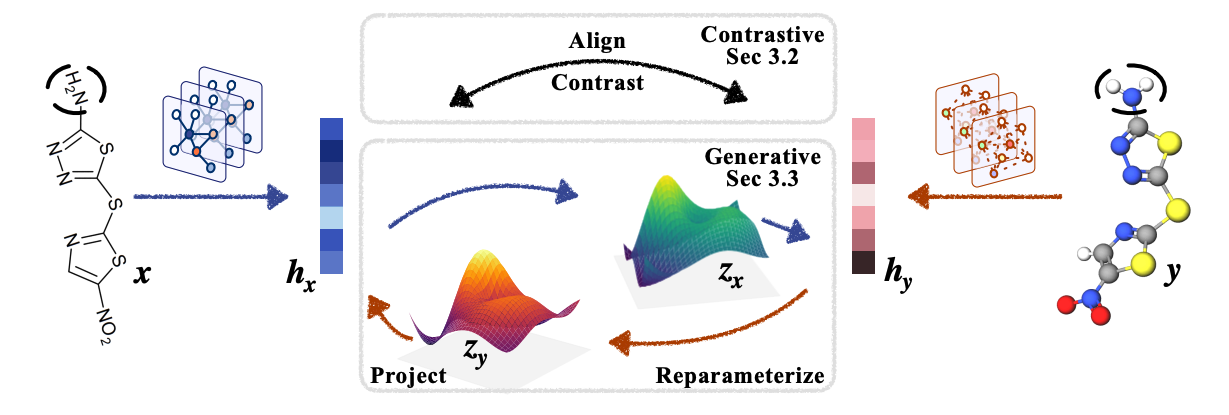
当我们有了公式1引出的一系列求解思路之后就非常直接。 GraphMVP是完全基于latent space,使用了如下两个目标函数: 一个contrastive loss,也就是EBM-NCE;一个generative loss,也就是VRR。除此以外,GraphMVP还有两个variant,GraphMVP-C和GraphMVP-G,分别考虑到了如何加入contrastive 和 generative 2D SSL。
2.2.3 基于data space的MoleculeSDE
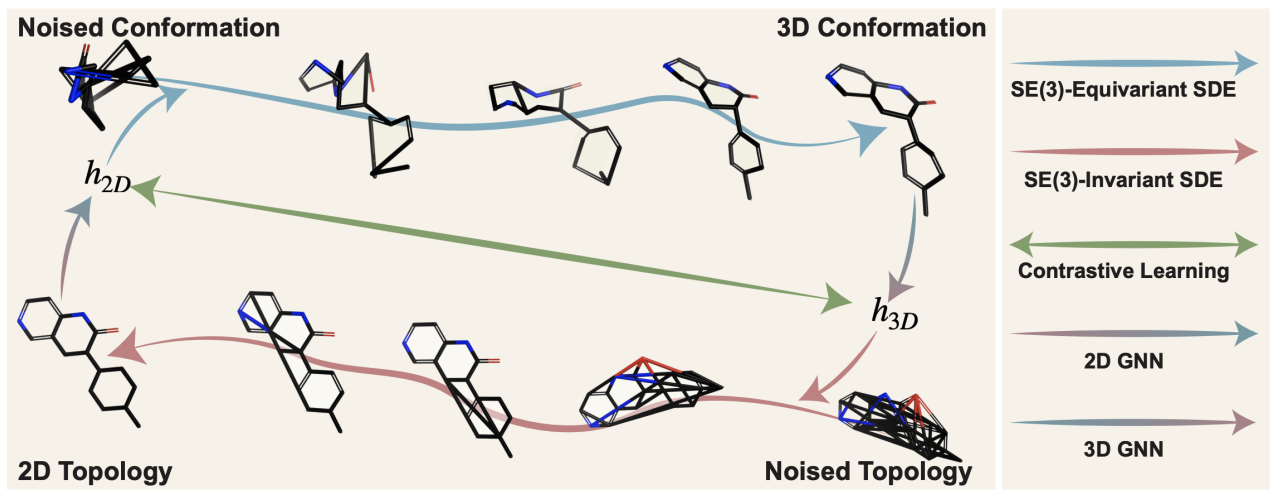
MoleculeSDE是GraphMVP的follow-up工作。在GraphMVP中的generative loss,我们是利用了VRR来对ELBO进行估计,但是这种估计会造成信息损失。这里我们提出了更加严格的概率估计,也就是直接在data space (geometry和topology)进行重构。
但这里又有一个挑战,就是从2D到3D的条件概率 (2D生成3D) 需要遵照SE(3)等变,即对于旋转、平移等变,并且对于对称反对称。为了实现这个目标,我们基于vector frame basis引入了SE(3)-equivariant and reflection-antisymmetric SDE。这个思路本质上是利用score matching或者diffusion去求解公式1。
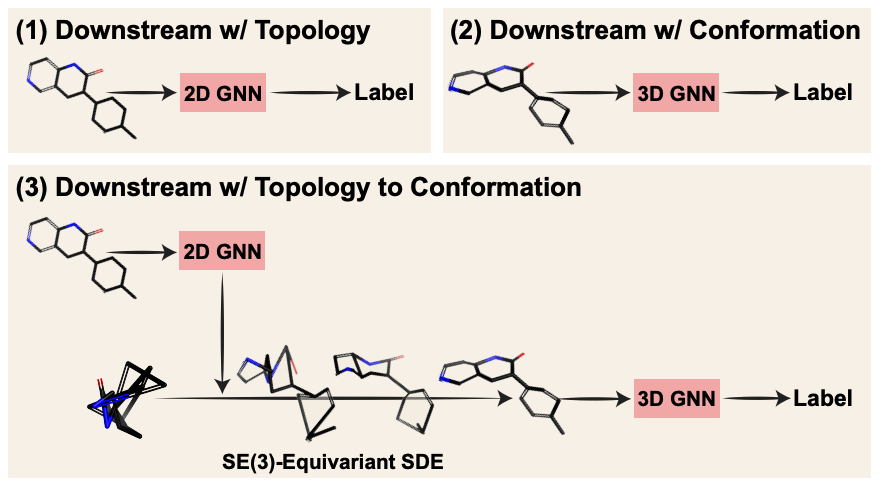
另外我们想强调的是,对于downstream task,除了常规的2D和3D proeprty prediction,MoleculeSDE也让我们有了更多样化的选择。主要是对于小分子2D到3D的生成,这个task的意义并不仅仅在于能够有conformation generation,而是能够有基于生成的3D coordinates 进一步进行modeling,如下图(3)所示:
2.3 GeoSSL: 3D pretraining
GraphMVP和MoleculeSDE都是考虑模态之间的预训练。同时我们还想强调仅仅考虑3D geometry的预训练。这个工作的推出是在GraphMVP和MoleculeSDE这两个工作之间,而原因则是因为预训练的数据集。GraphMVP的预训练是在GEOM数据上(当时取了250K data,是已知较大的小分子3D数据集)。而2021年暑假开始,陆续有几个比较大的数据集相继推出,比如Molecule3D和PCQM4Mv2。GeoSSL就是在Molecule3D上进行预训练。
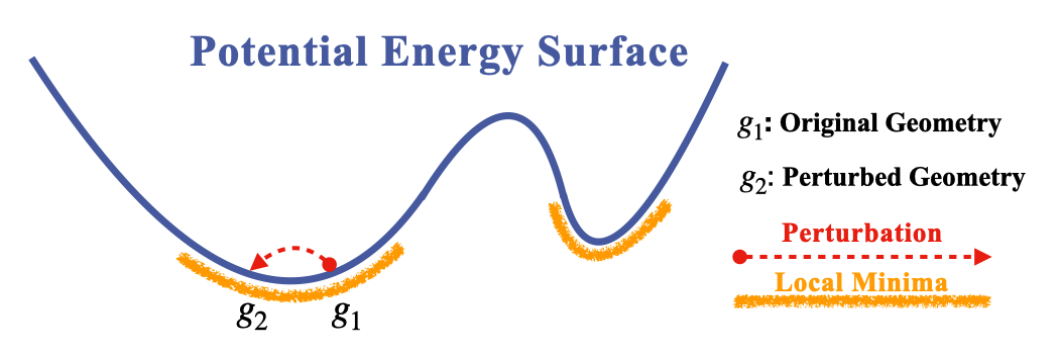
GeoSSL是仅仅考虑到geometry的single-modality pretraining。我们首先需要定义view。这里的出发点是在计算或者模拟中,分子的3D coordinates有一定的误差,并且分子的3D geometry 哪怕是稳定态(势能面),也会在一个小区域内进行运动。由此,我们定义了两个view:original geometry和perturbed geometry,如下图所示。
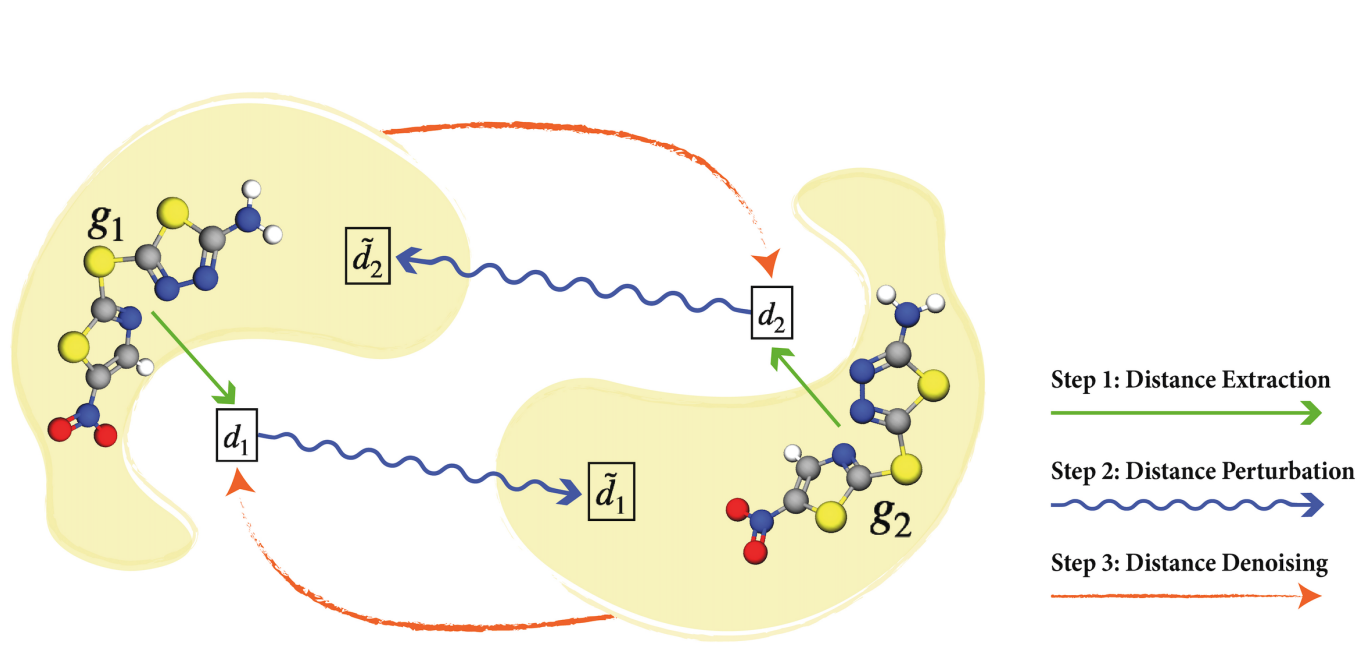
基于这两个view,我们又可以利用公式1引出的一系列方法来最大化MI。已有的方法 (比如EBM-NCE、InfoNCE、RR)都在GeoSSL中进行了benchmark。此外我们还利用了geometry数据的特性,也就是连续的3D coordinates,提出了利用denoising score matching的方法,来进行denoising distance matching,从而帮助了参数估计。具体推导过程可以看论文。大概流程则如下图所示:
2.4 小结
下图就是我们对于整个公式1的几种不同求解思路的roadmap:

分子geometry representation learning本身已经是一件挑战性同时非常重要的任务,因为geometry是这些物理粒子最本质的并且很复杂的数据结构;而geometry预训练任务的复杂性更加大一些。我们团队的工作一直在探索相关问题。从最开始的benchmark,一直到预训练,所有的代码除了每一个工作各自的github repo开源,也已经整合到了Geom3D这个平台上。
Chapter 3 textual description modality enables more comprehensive tasks
3.1 GraphCG 基于图的分子编辑(可控生成)
GraphCG是我们第一个使用深度学习来进行可控生成的工作。目前已有的可控生成主要是对于图片的操作,而我们认为,对于分子的可控生成,可以很好地用于lead optimization等药物发现的重要任务中。问题的出发点是说,现有的lead optimization都高度依赖于专家的经验,因此比较耗费人力并且有一定的主观性。而当我们如果能够成功利用深度学习的方法,挖掘出图生成模型中的可控因子,那么就能够提供另外一种lead optimization方法,从而帮助药物专家进行研发。
问题的设定是给定一个已经训练完成的图生成模型,然后我们主要进行了两个步骤的分析:
- 首先我们验证了已有的图生成模型,都在一定程度上是高度耦合的 (entangled)。因为高度耦合,所以要实现可控生成就比较有挑战性。
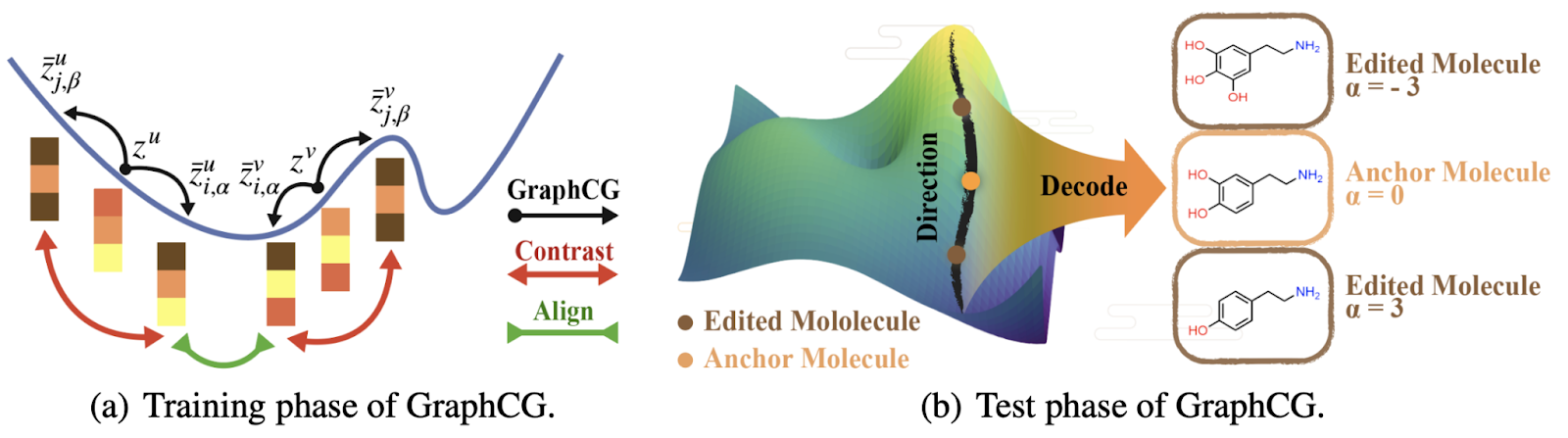
- 那么对于高度耦合的图生成模型 (已经训练完毕),如果我们想对它们进行可控生成,是否可行?答案是可以的,我们对于此提出了GraphCG。
- 在latent space中,我们有不同的semantic direction,而每一个semantic direction都有各自对应的特定的可控因子。
- 如果我们想学习到这些semantic direction,我们先有一个假设:不同的图,在latent space中如果能够沿着一个semantic direction移动,那么它们就会有对应的可控因子改变 (比如某一个图子结构变多)。这种假设可以通过最大化互信息来实现。这就回到了我们的公式1。具体求解的时候,我们利用了NCE的求解方案。这样我们就利用最大化互信息学到了这些semantic direction。
- 在inference过程中,我们只需要把每一个图对应的latent representation 沿着 semantic direction 进行移动,然后进行解码,这个解码之后的图就是我们希望的某一个因子改变的图。
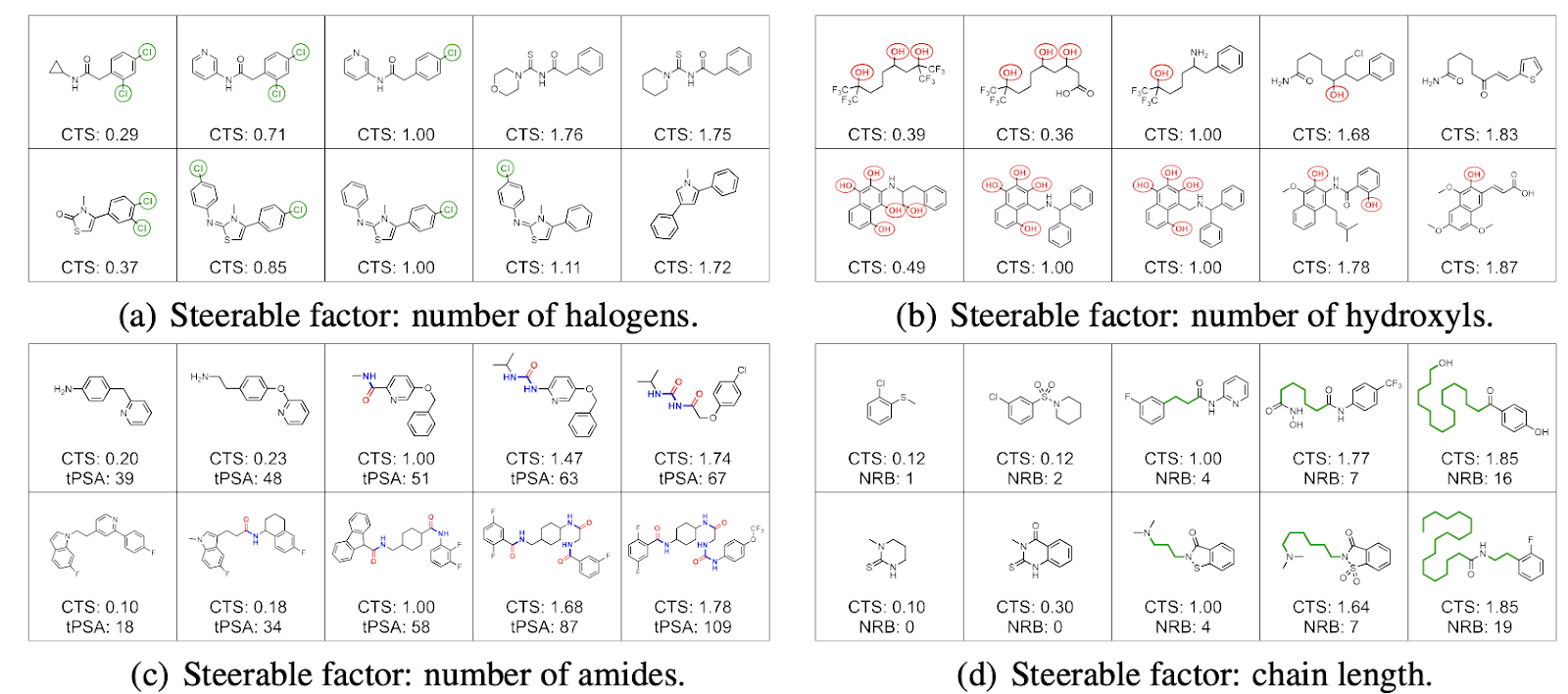
具体定量结果可以参考原文,这里我们主要展示定性结果。
首先在分子图,我们利用了已经训练好的HierVAE,在它的latent space上训练得到了10个semantic direction。然后我们发现其中四个semantic direction都能对应到专门的分子官能团。这个很好地符合了我们的期望,并且也能够帮助药物专家进行lead optimization设计:比如希望对原始分子图增加/减少更多的halogen基团,见下图(a)。
其次我们还展示了基于PointFlow的点云图的可控结果。下图中的a和b是一个方向,并且我们能看到往左往右两个不同的方向,对应的飞机引擎数目会分别减少和增加。此外在车子和椅子的外形也会随着可控因子,有对应的改变。

GraphCG的初步尝试给我们带来了非常大的信心,让我们对于分子的可控生成有了更大的把握。它也引导了我们后续的几个工作。
3.2 MoleculeSTM 基于文本的分子编辑
最近随着大模型、多模态的应用,一个很自然的想法就是我们是否也可以将这些技术用到药物发现上?并且这些自然语言的文本描述,是否对于药物发现这个有挑战性的问题带来新的视角?答案是肯定并且乐观的。
具体到方法上。MoleculeSTM的核心思路非常简单直接:分子的描述有内部化学结构和外部功能描述两大类,而我们这里利用了multi-modal learning的思路,将两种类型的信息进行联系,并且基于此我们设计了种类丰富的下游任务来验证其有效性。这里预训练的思路还是通过求解公式1来给两个模态(对应的表征函数)进行链接。
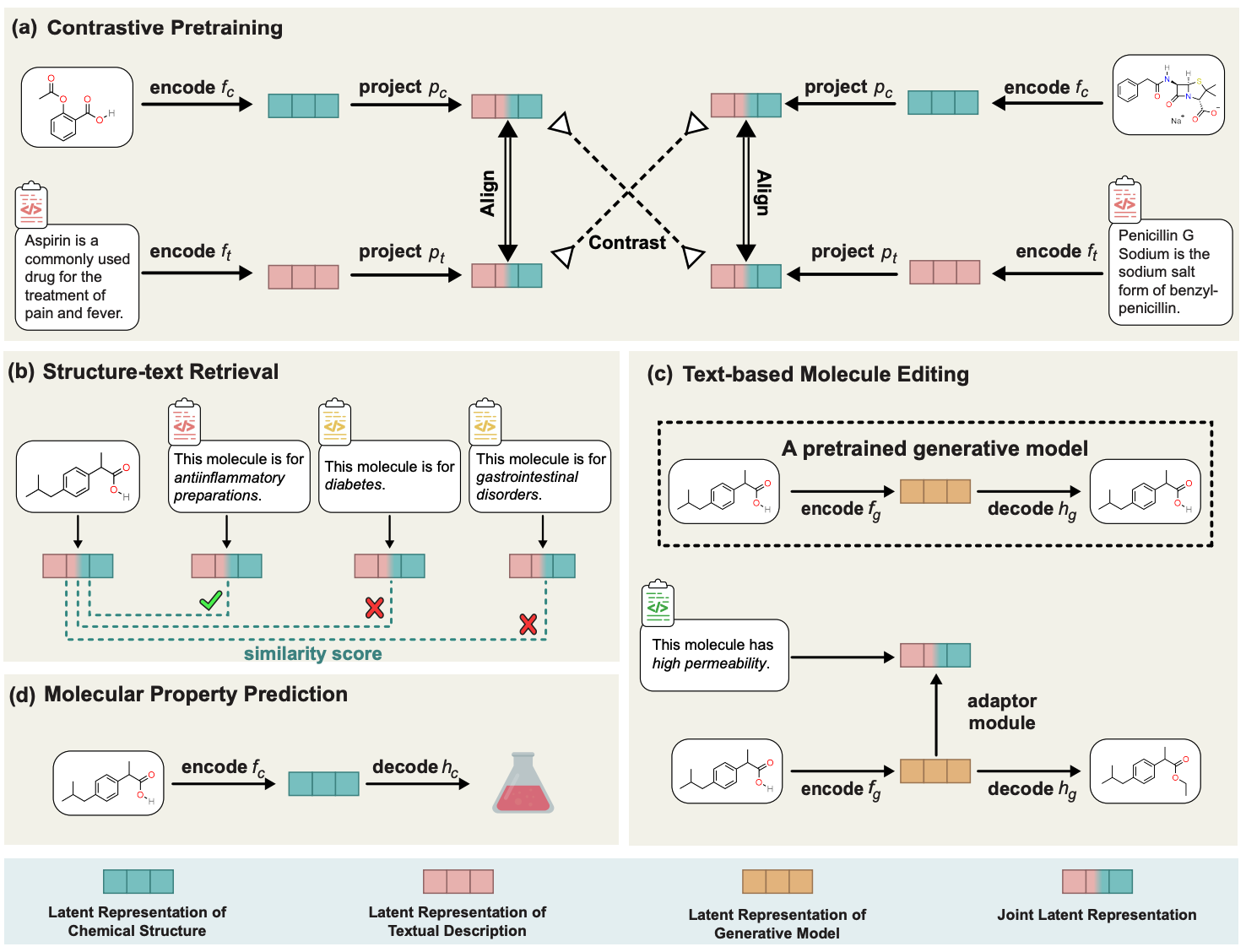
下面我们主要强调几个insight。
3.2.1 自然语言和大语言模型有什么特点能够帮助scientific discovery?
这个是我们在MoleculeSTM中首先提出的问题。在MoleculeSTM这个工作里,我们是利用了自然语言的open vocabulary和compositionality特性。
- open vocabulary是说我们对于药物的描述可以非常的多样,比如一个极端例子是“开发一个药物能够治疗明年的突发的xxx疾病”这样的text prompt,只不过这种text prompt是难以验证,并且对于目标的描述过于模糊。
- compositionality是说我们有一些比较复杂的多目标任务,但是如果在自然语言中,它更容易组合。一个例子是多属性的lead optimization,也就是让一个分子优化到同时符合多个属性;传统做法会需要对每一个属性训练一个分类器,而MoleculeSTM仅仅只需要把两个属性用自然语言描述然后通过“and”连接即可。
- 在我们最近的工作ChatDrug中,我们又挖掘了自然语言和大语言模型的对话特性。这个会在下面展开。
3.2.2 自然语言和大语言模型的特点能够帮助什么类型的scientific discovery任务?
现有的language-vision task可以认为是艺术相关的任务 (比如生成图片、文字),也就是说它们的结果是可以多样和不确定。但是scientific discovery是科学问题,通常有着比较明确的结果,比如生成有某个功效的小分子。这个在任务的设计上带来了更大的挑战。
在MoleculeSTM中 (Appendix B),我们提出了两个准则:
- 首先我们考虑的任务是能够进行计算模拟得到结果。将来会考虑能够有wet-lab验证的结果,但这并不在目前这个工作的考量范畴内。
- 其次我们只考虑有着模糊性描述的问题(问题的描述是相对模糊的,但是答案是相对确定)。具体例子比如让某个分子的水溶性或者穿透性变强。而有一些问题有明确结果,比如在分子的某一个位置加入某一个官能团,我们认为这类任务对于药物、化学专家来说更加简单直接,而DL的帮助比较有限;所以它可以将来当作某一个proof-of-concept任务,但是并不会成为主要的任务目标。
3.2.3 分子编辑的定性结果
MoleculeSTM的文章中我们引入了三类下游任务,来验证MoleculeSTM的有效性。这里我们主要想强调一下zero-shot text-guided molecule editing的定性结果。
这个task就是同时输入一个分子和自然语言描述(比如额外的属性),然后希望能够输出复合语言文本描述的新的分子。这也是text-guided lead optimization,一种新的lead optimization范式。
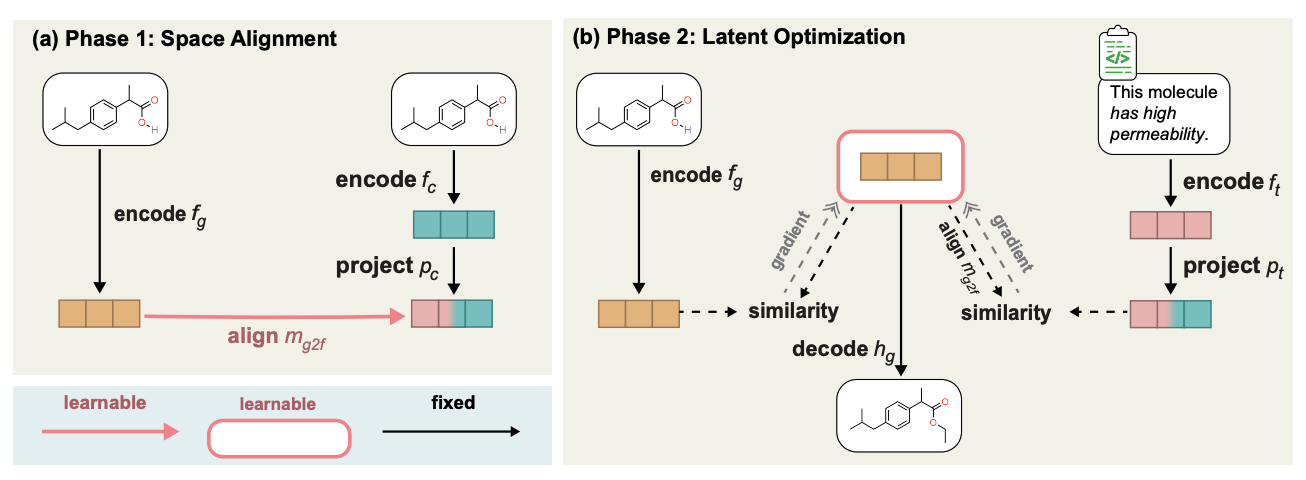
具体的方法就是利用已经训练好的分子深成模型和我们预训练好的MoleculeSTM,通过学习二者的latent space的alignment,从而进行 latent space interpolation,再经过解码生成目标分子。流程示意图如下。
这里我们展示了几组分子编辑的定性结果 (其余下游任务的结果细节可以参考原论文)。主要我们考虑五类分子编辑任务:
- 单一属性编辑:对单一属性进行编辑,比如水溶性、穿透性、氢键施主与受主个数。
- 复合属性编辑:同时对多个属性进行编辑,比如水溶性和氢键施主个数。
- 药物相似性编辑:(Appendix D.5)是让输入分子与目标分子药物长得更加接近。
- 专利药物的邻居搜索:对于已经申请到专利的药物,往往会把中间过程的药物一起报道。我们这里就是那中间药物配合自然语言描述,看是否能够生成最终的目标药物。
- binding affinity编辑:我们选择几个ChEMBL assay作为target,目标是让输入分子和target有更高的binding affinity。
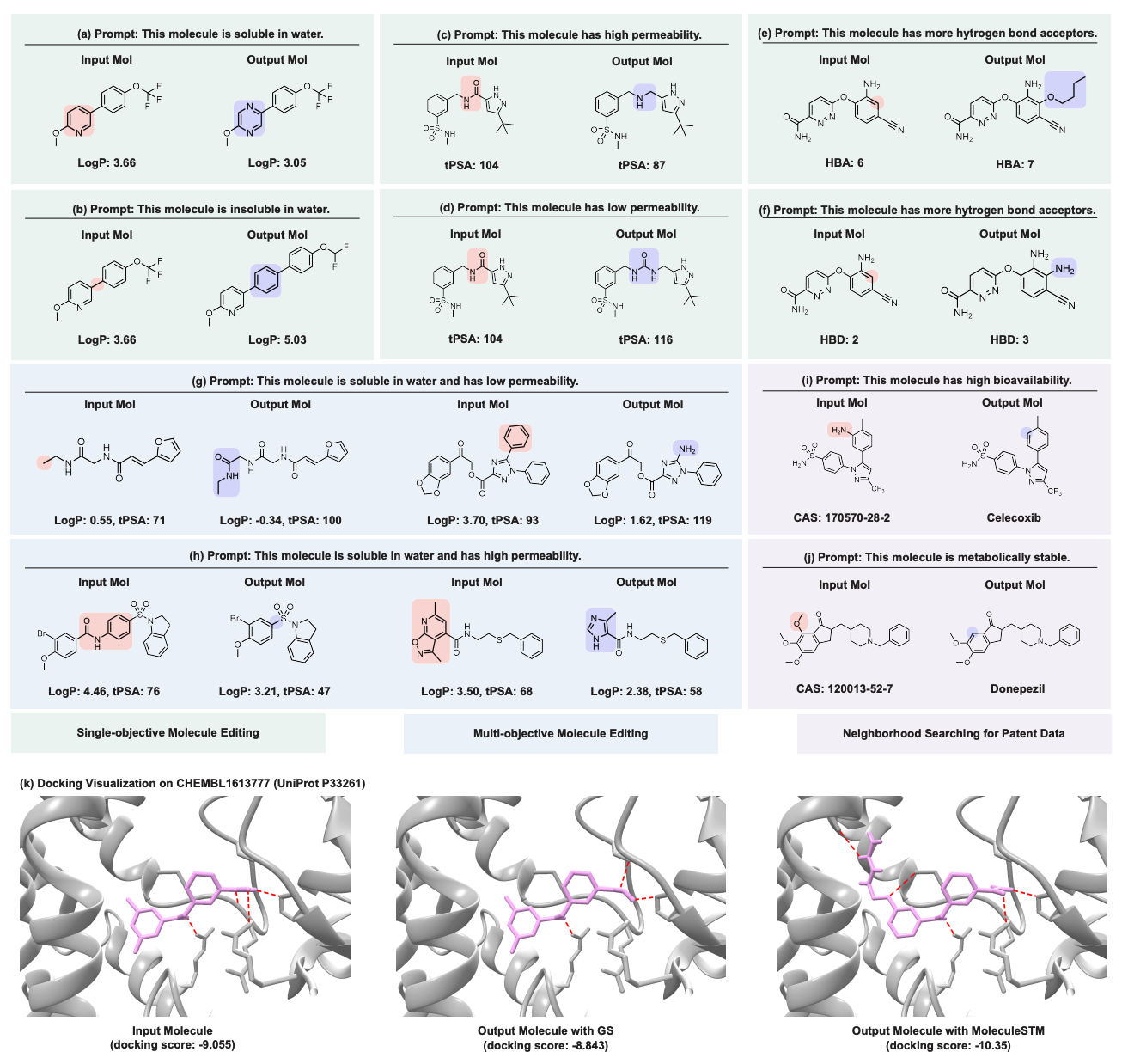
3.3 ProteinDT 基于文本的蛋白质生成和编辑
MoleculeSTM的初步探索让我们感受到文本描述的潜在用途是能够提供更多解决问题的视角。这个工作我们就很自然的将这个想法拓展到蛋白质领域上。
首先在预训练上,ProteinDT主要分为三步:
- 第一步ProteinCLAP,是将text sequence和protein sequence通过contrastive learning来学习alignment。
- 第二步Facilitator,是进一步增强alignment。
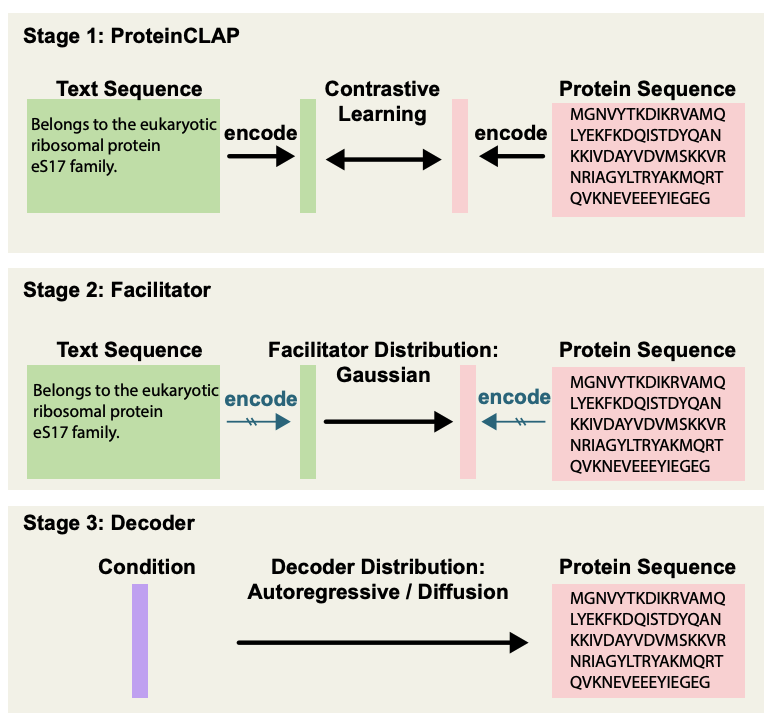
- 前两个步骤都是通过latent space操作,而第三步deconder则是将latent representation解码到data space。需要提到的是我们这里考虑了两类decoder:auto-regressive和denoising diffusion(如下图)。
- 这里我们还想要强调的是,Transformer并不是生成模型,而仅仅是一个深度学习模块。我们的两种生成模型都考虑了用Transformer作为核心模块的情况。
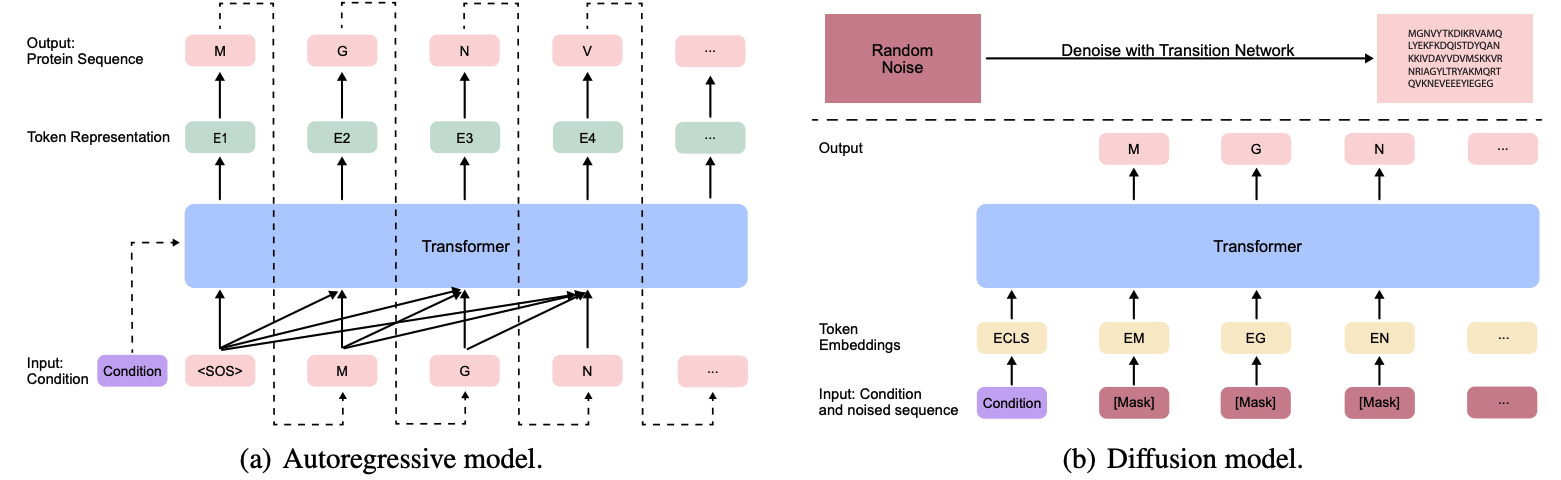
3.3.1 Text-to-Protein Generation
当我们有了上述的流程图之后,就可以进行text-to-protein generation的生成(如下图)。并且我们还进行了消融实验证明了facilitator模块的必要性。
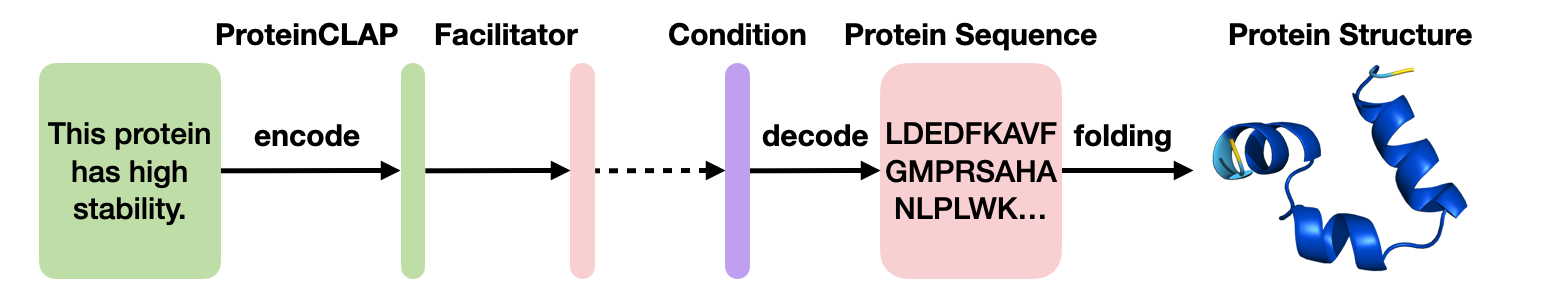
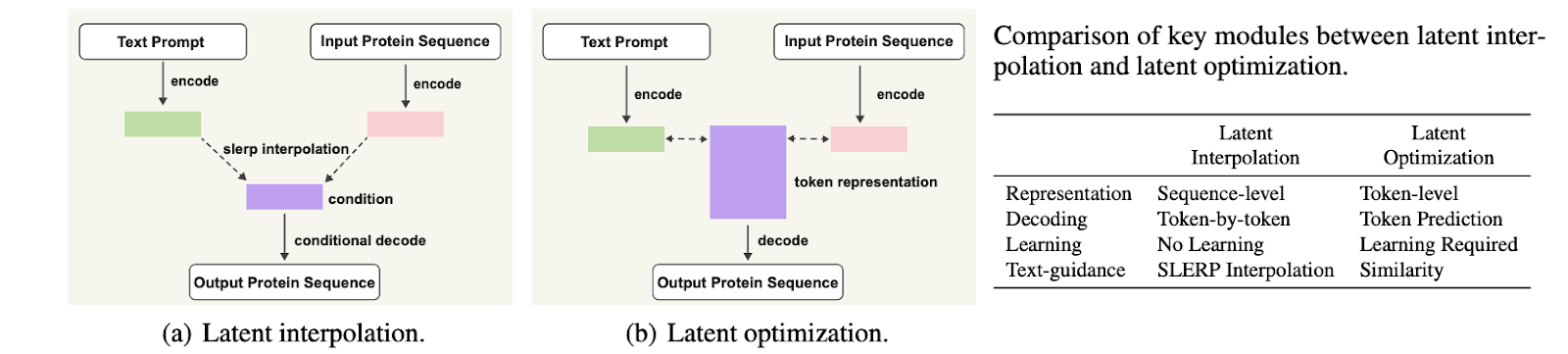
3.3.2 Text-guided Protein Editing
这里基于ProteinCLAP,我们提出了两种protein editing思路。
- latent interpolation是直接在latent space进行控制interpolation,然后直接解码到protein sequence space。
- latent optimization则是专门训练一个token-level的解码器,然后利用ProteinCLAP直接优化得到optimal latent,再通过解码到protein sequence space。
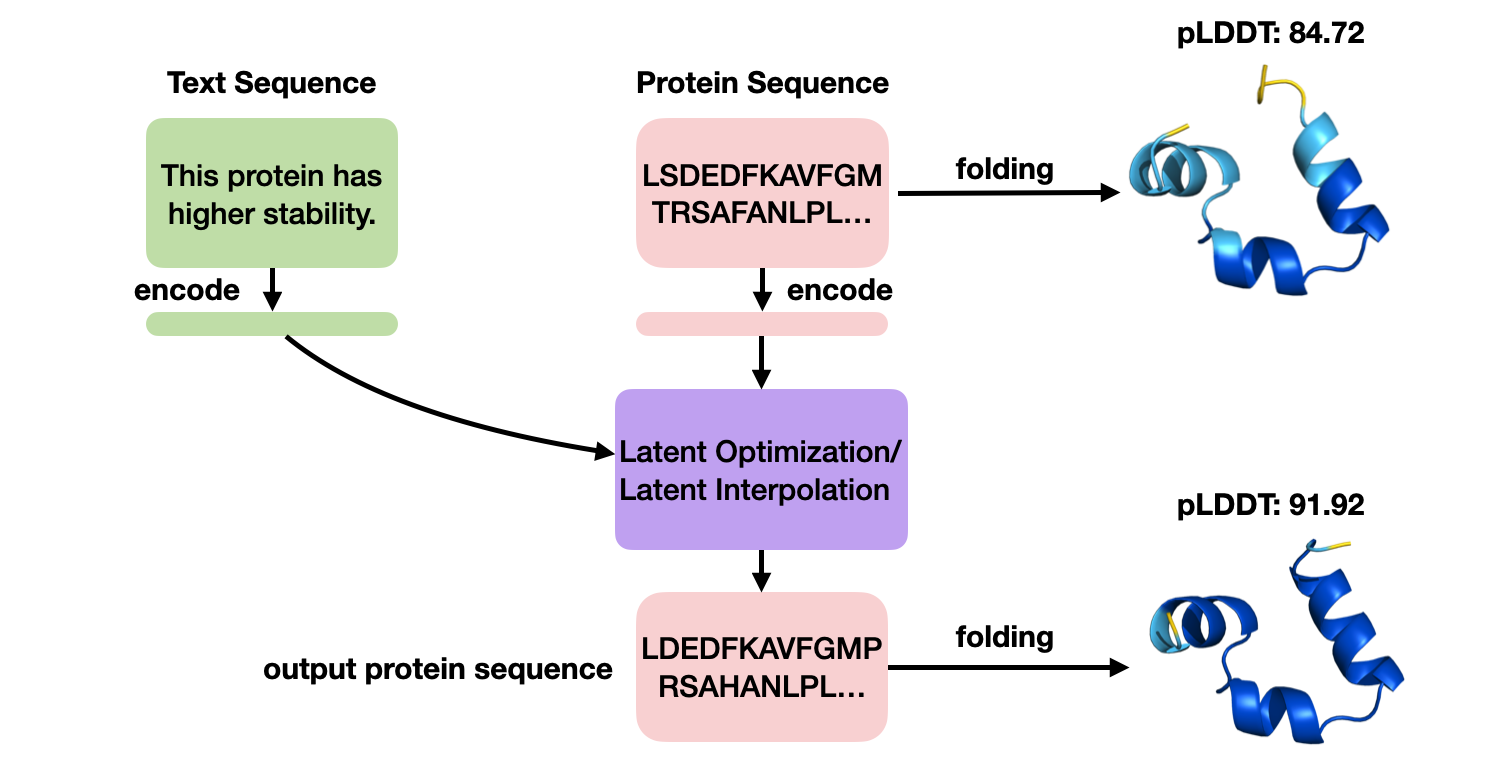
整个text-guided protein editing的inference流程图如上图。更多定量实验结果可以参考原文。
3.4 ChatDrug 基于ChatGPT 对话增强的小分子、多肽、蛋白质编辑
在2022年的11月,ChatGPT推出。紧接着很多领域、应用都开始尝试使用这个基于大模型的工具,并且发现了它的确能够优雅地解决很多问题。
这个工作中,我们就尝试将ChatGPT用于药物发现。首先一个比较有挑战性的瓶颈还是如何设计任务。我们跟随MoleculeSTM和ProteinDT的insight,主要对标drug editing系列任务,并且我们在这里考虑了三种类型的药物:小分子、多肽、蛋白质。
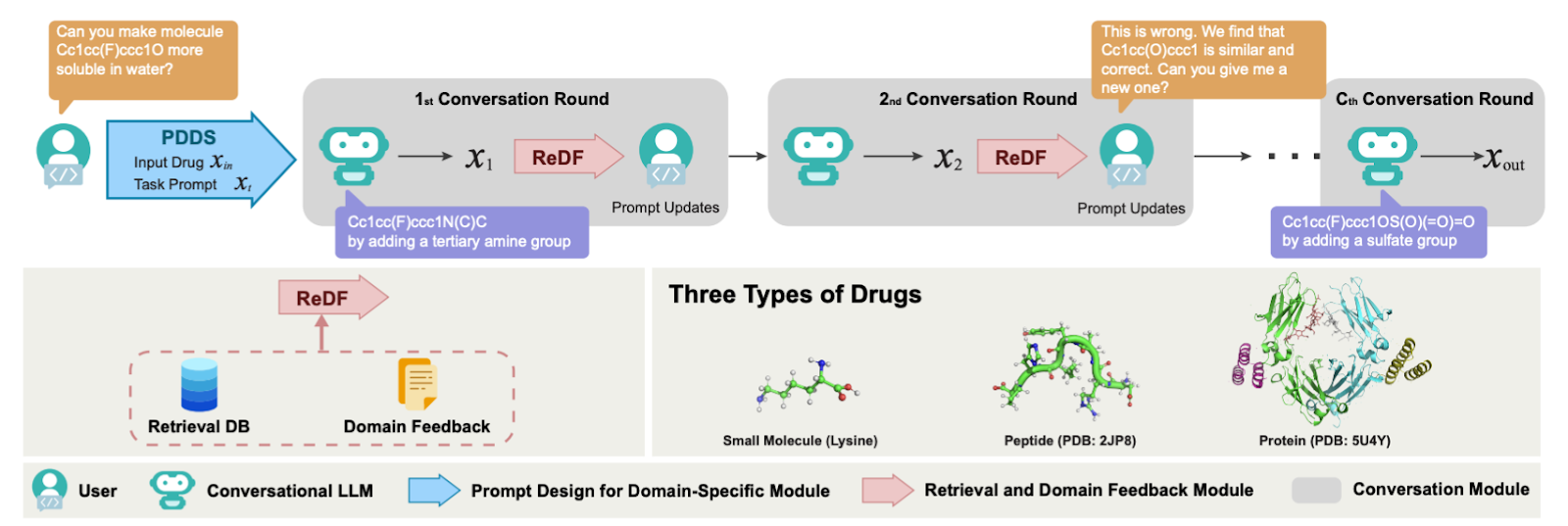
根据ChatGPT的特性,我们提出了ChatDrug,它主要有三个模块:
- PDDS模块是针对domain task设计的prompt。
- ReDF模块是利用retrieval和domain feedback对prompt进行更新。
- conversation module是让用户和ChatDrug进行交互,从而不断更新结果。
下面我们列出主要的定性结果。关于更详细的任务设定和定量结果,感兴趣的朋友可以看文章细节。
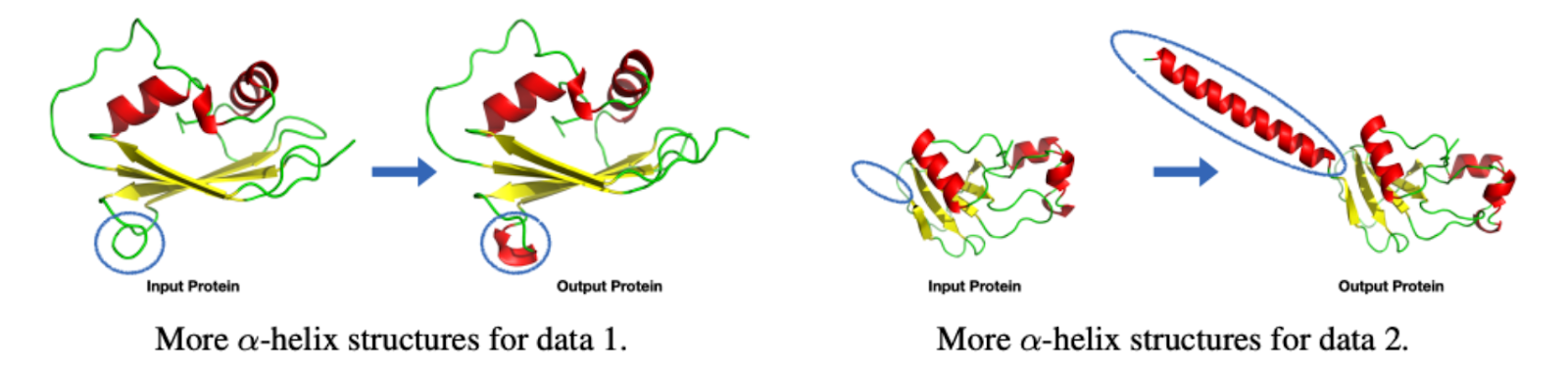
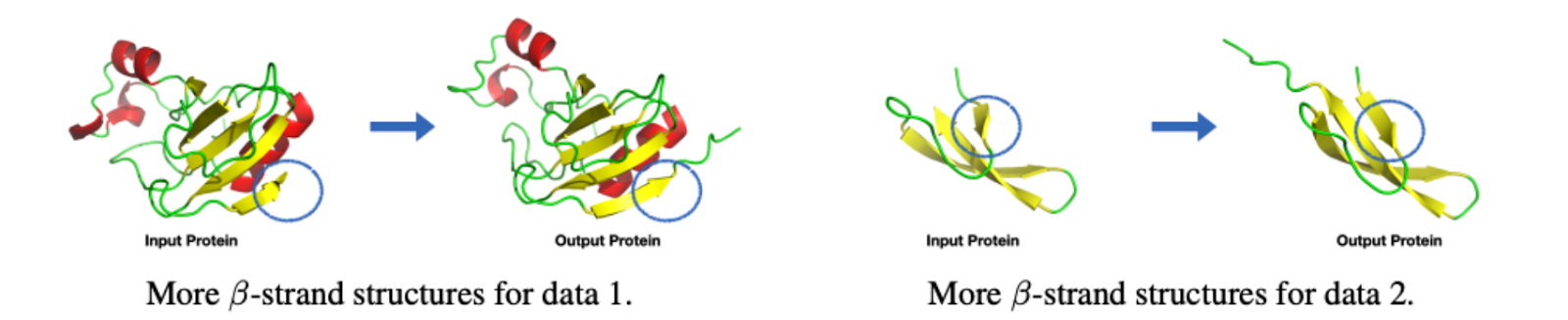
- ChatDrug 小分子编辑定性结果:
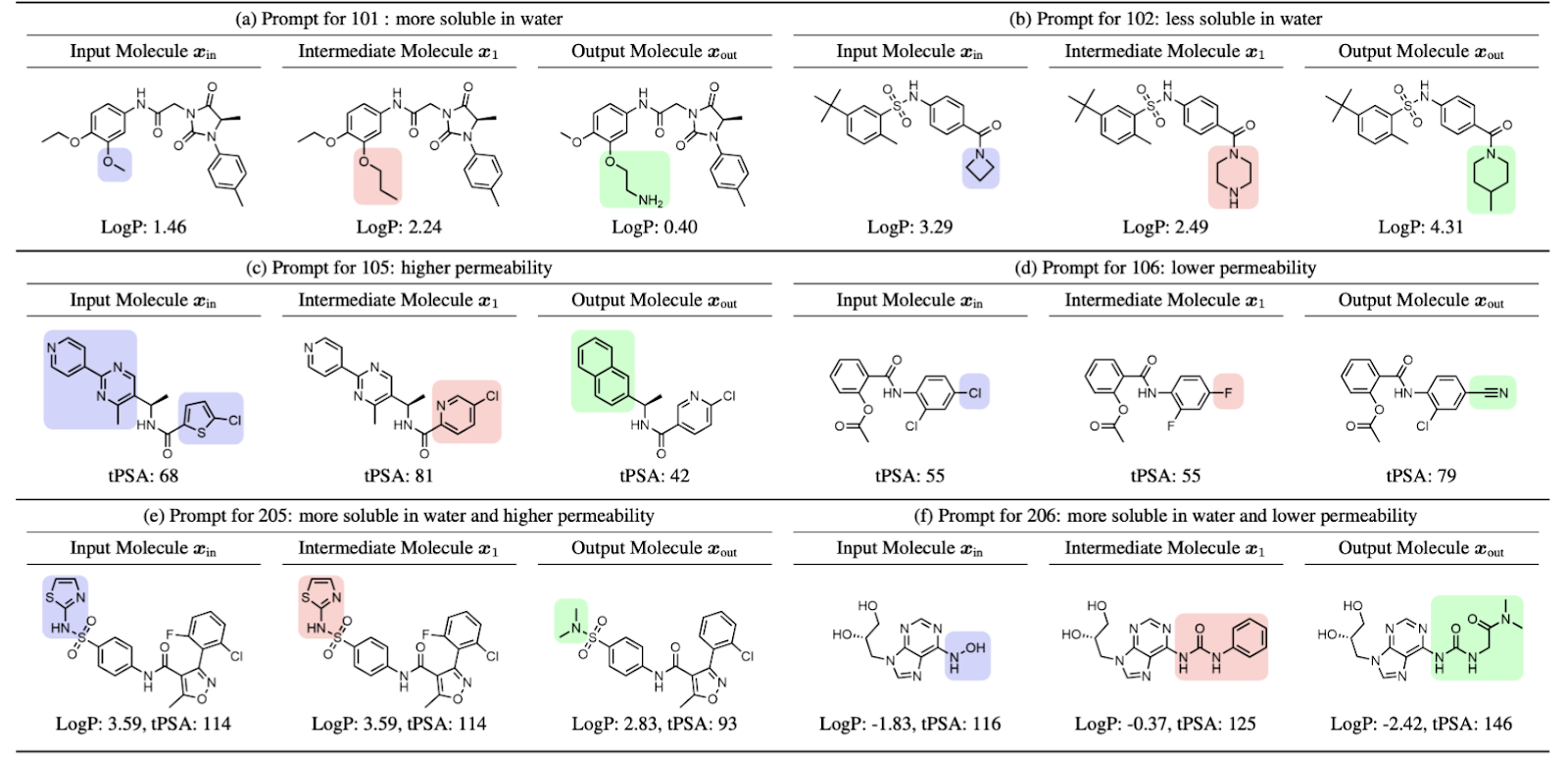
- ChatDrug 多肽编辑定性结果:

- ChatDrug 蛋白质编辑定性结果:
Chapter 4 总结
我们通过前面两个系列从ML技术和domain解释两个角度介绍了分子的多模态任务。此外我们团队还有一些其他的探索,和前面两个系列的工作一起在下图展示。
整个AI for molecule/drug discovery的发展其实还是比较初步,但是已经吸引到学术界、工业界的大量关注。回首十年前深度学习刚开始随着GPU的发展影响学术圈,而十年后的现在深度学习在艺术领域已经取得了非常大的突破。我们团队也是对深度学习在科学领域的前景很有信心,但这两个领域之间目前也存在着非常大的gap:
- 对于domain科学家,往往把深度学习当做一个可以直接使用解决问题的一个黑盒模块,但是这个操作往往忽略了重要的优化过程。这个可以直接通过理解优化过程(目前有一些物理统计的尝试,但还是非常困难),或者目前更加可操作的就是更严格的计算上的控制变量实验。
- 而人工智能领域的科研工作者往往考虑一些比较简单的评估方法和任务。这个就可以多去和domain专家沟通、理解domain上的问题。
这些挑战是交叉领域发展的自然过程,下面的一个方向就需要数学家、物理学家、统计学家、化学家、生物学家、工程师等各个领域/技能的科研学者进行更多深入的交流合作,构造起一个更加严格的pipeline。一个非常好的例子就是AlphaFold,它在兼顾对解决folding问题有突破性进展的同时,又有非常深厚的数学物理支撑,并且对solvable的问题敏锐程度非常精准。希望将来能有更多这样的工作出现。
- [1] GraphMVP: Pre-training Molecular Graph Representation with 3D Geometry, ICLR 2022
- [2] GeoSSL: Molecular Geometry Pretraining with SE(3)-Invariant Denoising Distance Matching, ICLR 2023
- [3] MoleculeSDE: A Group Symmetric Stochastic Differential Equation Model for Molecule Multi-modal Pretraining, ICML 2023
- [4] Geom3D: Symmetry-Informed Geometric Representation for Molecules, Proteins, and Crystalline Materials, ArXiv 2023
- [5] GraphCG: Unsupervised Discovery of Steerable Factors in Graphs, NeurIPS Workshop 2022
- [6] MoleculeSTM: Multi-modal Molecule Structure-text Model for Text-based Editing and Retrieval, ArXiv 202
- [7] ProteinDT: A Text-guided Protein Design Framework, ArXiv 2023
- [8] ChatDrug: ChatGPT-powered Conversational Drug Editing Using Retrieval and Domain Feedback, ArXiv 2023
- [9] N-Gram Graph: Simple Unsupervised Representation for Graphs, with Applications to Molecules, NeurIPS 2019
- [10] AWARE: Attentive Walk-Aggregating Graph Neural Networks, TMLR 2022
- [11] SGNN-EBM: Structured Multi-task Learning for Molecular Property Prediction, AISTATS 2022
- [12] GIMLET: A Unified Graph-Text Model for Instruction-Based Molecule Zero-Shot Learning, ArXiv 2023
- [13] MolGraphEval: [Evaluating Self-Supervised Learning for Molecular Graph Embeddings, ArXiv 2022](https://arxiv.org/abs/2206.08005_
- [14] D3G: Leveraging Domain Relations for Domain Generalization, ArXiv 2023
致谢
这个博客是我对于过去三年半工作的一个简短的总结。这里两个系列中,最早的工作是GraphMVP和Geom3D,都是21年暑假开始的工作。
首先非常感谢各位指导老师的大力支持。Chapter 2是我主要在Mila Ph.D.期间完成的工作,非常感谢唐建教授的指导。Chapter 3主要是我在Caltech & Nvidia Prof. Anima Anandkumar指导下完成,她的指导非常有远见。此外也非常感谢加拿大国家实验室的Prof. Hongyu Guo还有UC Berkeley的Prof. Omar Yaghi, Prof. Christian Borgs, Prof. Jennifer Chayes的支持。
我还十分感谢所有合作者的参与讨论,尤其是UIUC的王程鹏博士,中科院的杜伟韬博士后,HKU的刘琦教授,ASU的Chaowei Xiao教授,斯坦福的王瀚宸博士后,UofT的李卓鑫燃同学。